Pairwise alignments of each species to the mouse genome are displayed as a grayscale density plot (in pack mode) or as a wiggle (in full mode) that indicates the proportion of matching bases in the target species. In squish mode, the number of matching bases in all species is shown as a single wiggle track.
Checkboxes on the track configuration page allow selection of the species to include in the pairwise display.
To view detailed information about the alignments at a specific position, zoom the display in to 30,000 or fewer bases, then click on the alignment.
Codon translation is available in base-level display mode if the displayed region is identified as a coding segment. To display this annotation, select the species for translation from the pull-down menu in the Codon Translation configuration section at the top of the page. Then, select one of the following modes:
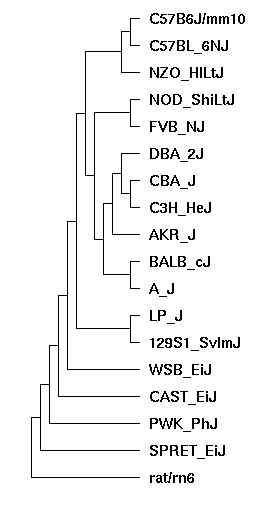
For this assembly hub, a Progressive Cactus alignment was generated using the Genbank version of these assemblies. The reference mouse (mm10) as well as the reference rat (rn6) were included as well. Here is the guide tree for this alignment:
Due to the highly similar nature of the laboratory mice strains, this tree was binarized as accurately as possible, but incomplete lineage sorting (ILS) is prevalent and as a result the guide tree may not be correct in all regions. This has minimal effect on alignment quality except in regions where insertions and deletions are under ILS.
The alignment underlying this track is also available here in HAL format. Due to load times problems with HAL files, the HAL file was used to generate the underlying bigMaf files for this track and is not directly part of the hub.
Alignment generation: Joel Armstrong, Ian Fiddes, Benedict Paten.
Genome assemblies: Thomas Keane, The Mouse Genomes Project.
Paten et al. Cactus: Algorithms for genome multiple sequence alignment. Genome research. 2011;21:1512-1528.
Nguyen et al.Comparative Assembly Hubs: Web Accessible Browsers for Comparative Genomics. Bioinformatics. 2014 Aug (advance online publication).Hickey et al. HAL: a hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics. 2013 May;29(10):1341-1342.
For general questions about these data, please contact tk2@sanger.ac.uk or ifiddes@ucsc.edu.