|
Can I visualize RNA-SEQ data on the Repeat Browser?
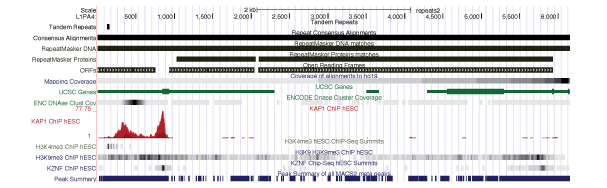
It is possible to map coverage tracks or other aspects of your RNA-SEQ data to the Repeat Browser, but it's not really clear what this means. The consensuses we provide have unequal coverage from all their genomic instances (see "Mapping Coverage" track in "Mapping to the Human Genome"). In simpler terms, if you look at all L1PA elements, there are many, many more bits of the 3' end scattered throughout the genome then full length elements so some sort of normalization would be needed to meaningfully interpret results mapped to the consensus.
If you are interested in quantifying the expression of TE families we recommend using existing tools designed for that purpose. TETranscripts, REdiscoverTE, and SalmonTE all do this in slightly different ways and can tell you if TEs are expressed in your dataset.
Can I visualize non-reference TE insertions on the Repeat Browser?
Depends on what you mean. Liftover files only exist for hg19 and hg38, so non-reference TEs don't have pre-computed coordinates on our consensuses and therefore can't be lifted. You can generate your own liftOver file for another assembly. Instructions on how to do that coming soon!
What you can also do is BLAT your TEs to the references and visualize them, similar to what we did for non-human primate TEs in our manuscript.
1) Download BLAT.
2) Then download the consensus sequence you wish to map to (you can View DNA for the element of interest in the browser, download the sequences from the Table Browser or download this file. You can also map to all the consensus sequences. Note mapping to all consensus sequences is slightly different than what we do when lifting. That is we only ever lift from an annotated L1PA2 to the L1PA2 consensus. If you blat a list of L1PA2 sequences against all consensuses (repeats2) you will almost certainly get mappings to other L1PAs. Be sure that's what you want to do.
3) BLAT them to create a psl file.
4) Load the psl as a custom track.
Are your references the RepBase consensuses?
No. Our consensuses are generated from RepeatMasker output. RepBase consensuses are available from GIRI with a subscription.
Why is <favorite TE element> not included?
Your TE probably isn't in the hg19 RepeatMasker output. Is your TE present in humans? If so, it's probably one of the TEs only present in hg38 annotations; if there is sufficient interest in it let us know we can try adding the consensus in a later update, but only hg38 data would be able to lift to it.
Where is L1PA9?
Good question, it's not in RepeatMasker output!
What is the deal with L1PA3long and L1PA3short?
Each of these are subclasses of L1PA3 elements. L1PA3 elements deleted a 129-bp region of themselves to escape ZNF93-mediated repression. See Jacobs et al, 2014 for details. Please note that the provided liftover files only lift to L1PA3long which is the L1PA3 consensus produced through our standard workflow.
Does this exist for mouse (mm10)?
No. Would you find a mouse browser useful? Let us know!
I have a question! I guess it's not frequently asked.
Please email
jferna10 at ucsc.edu
max at soe. ucsc.edu
|